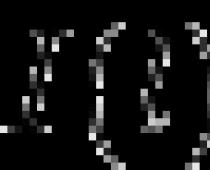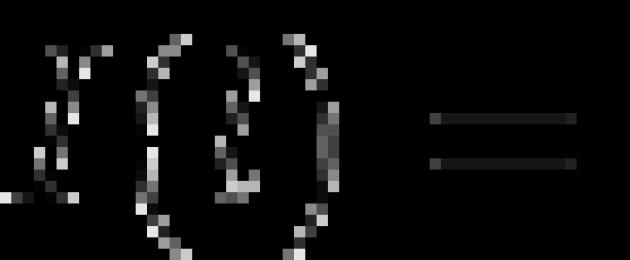
Введение
Глава 1. Общие понятия. Классификация электрохимических методов анализа
Глава 2. Потенциометрические методы анализа (потенциометрия)
1 Принцип метода
3 Потенциометрическое титрование
Глава 3. Кондуктометрический метод анализа
1 Принцип метода. Основные понятия
2 Принцип кондуктометрии
3 Кондуктометрическое титрование
Глава 4. Кондуктометрический анализ (кондуктометрия)
1 Сущность метода
2 Количественный полярографический анализ
3 Применение полярографии
Глава 5. Амперометрическое титрование
Глава 6. Кулонометрический анализ (кулонометрия)
1 Принцип метода
3 Кулонометрическое титрование
Заключение
Список литературы
ВВЕДЕНИЕ
Электрохимические методы анализа - это совокупность методов качественного и количественного анализа, основанных на электрохимических явлениях, происходящих в исследуемой среде или на границе раздела фаз и связанных с изменением структуры, химического состава или концентрации анализируемого вещества.
Электрохимические методы анализа делятся на пять основных групп: потенциометрию, вольтамперометрию, кулонометрию, кондуктрометрию и амперометрию.
Применение данных методов в количественном анализе основано на зависимости величин измеряемых параметров при протекании электрохимического процесса от отделяемого вещества в анализируемом растворе, участвующем в данном электрохимическом процессе. К таким параметрам можно отнести разность электрических потенциалов, количество электричества. Электрохимические процессы - это процессы, которые одновременно сопровождаются протеканием химической реакции и изменением электрических свойств системы, которую в подобных случаях можно назвать электрохимической системой. В аналитической практике, электрохимическая система обычно содержит электрохимическую ячейку, включающую сосуд с электропроводящим анализируемым раствором, в который погружены электроды.
Различают прямые и косвенные электрохимические методы. В прямых методах используют зависимость силы тока (потенциала и тому подобное) от концентрации определяемого компонента. В косвенных методах силу тока (потенциал и тому подобное) измеряют с целью нахождения конечной точки титрования определяемого компонента подходящим титрантом, то есть используют зависимость измеряемого параметра от объема титранта.
ГЛАВА 1. ОБЩИЕ ПОНЯТИЯ. КЛАССИФИКАЦИЯ ЭЛЕКТРОХИМИЧЕСКИХ МЕТОДОВ АНАЛИЗА
Электроаналитическая химия включает электрохимические методы анализа, основанные на электродных реакциях и на переносе электричества через растворы.
Применение электрохимических методов в количественном анализе базируется на использовании зависимостей величин измеряемых параметров электрохимических процессов (разность электрических потенциалов, ток, количество электричества) от содержания определяемого вещества в анализируемом растворе, участвующего в данном электрохимическом процессе. Электрохимические процессы - такие процессы, которые сопровождаются одновременным протеканием химических реакций и изменением электрических свойств системы, которую в подобных случаях можно назвать электрохимической системой. В аналитической практике электрохимическая система обычно содержит электрохимическую ячейку, включающую сосуд с электропроводящим анализируемым раствором,в который погружены электроды.
Классификация электрохимических методов анализа. Электрохимические методы анализа классифицируют по-разному.Классификация, основанная на учете природы источника электрической энергии в системе. Различают две группы методов:
а) Методы без наложения внешнего (постороннего) потенциала.
Источником электрической энергии служит сама электрохимическая система, представляющая собой гальванический элемент (гальваническую цепь). К таким методам относятся потенциометрические методы. Электродвижущая сила - ЭДС - и электродные потенциалы в такой системе зависят от содержания определяемого вещества в растворе.
б) Методы с наложением внешнего (постороннего) потенциала. К таким методам относятся:
кондуктометрический анализ - основан на измерении электрической проводимости растворов как* функции их концентрации;
вольтамперометрический анализ - основан на измерении тока как функции приложенной известной разности потенциалов и концентрации раствора;
кулонометрический анализ - основан на измерении количества электричества, прошедшего через раствор, как функции его концентрации;
электрогравиметрический анализ - основан на измерении массы продукта электрохимической реакции.
Классификация по способу применения электрохимических методов. Различают прямые и косвенные методы.
а)Прямые методы. Измеряют электрохимический параметр как известную функцию концентрации раствора и по показанию соответствующего измерительного прибора находят содержание определяемого вещества в растворе.
б)Косвенные методы - это методы титрования, в которых окончание титрования фиксируют на основании измерения электрических параметров системы.
В соответствии с данной классификацией различают, например, прямую кондуктометрию и кондуктометрическое титрование.
ГЛАВА 2. ПОТЕНЦИОМЕТРИЧЕСКИЙ МЕТОД АНАЛИЗА (ПОТЕНЦИОМЕТРИЯ)
1 Принцип метода
Потенциометрический анализ (потенциометрия) основан на измерении ЭДС и электродных потенциалов как функции концентрации анализируемого раствора.
Если в электрохимической системе - в гальваническом элементе -на электродах протекает реакция:
аА+bВ↔dD + еЕ
с переносом п электронов, то уравнение Нернста для ЭДС Е этой реакции имеет вид:
E꞊E˚- RTnFlnaDda Eea(A)a aBb
где, как обычно, Е° - стандартная ЭДС реакции (разность стандартных электродных потенциалов), R - газовая постоянная, Т - абсолютная температура, при которой протекает реакция, F - число Фарадея; а(А), a(В), a(D) и я(Е) - активности реагентов - участников реакции. Уравнение (10.1) справедливо для ЭДС обратимо работающего гальванического элемента.
Для комнатной температуры уравнение (10.1) можно представить в форме:
E꞊E˚- 0,059nlnaDda Eea(A)a aBb
В условиях, когда активности реагентов приблизительно равны их концентрации, уравнение (1) переходит в уравнение (3):
꞊E˚- RTnFlncDdc EecAa aBb
где с(А), с(В), с(Е), c(D) - концентрации реагентов. Для комнатной температуры это уравнение можно представить в виде (4):
꞊E˚- 0,059nlncDdc EecAa aBb
При потенциометрических измерениях в электрохимической ячейке используют два электрода: индикаторный электрод, потенциал которого зависит от концентрации определяемого (потенциалопределяющего) вещества в анализируемом растворе, и электрод сравнения, потенциал которого в условиях проведения анализа остается постоянным. Поэтому величину ЭДС, определяемую уравнениями (1)-(4), можно рассчитать как разность реальных потенциалов этих двух электродов.
В потенциометрии используют электроды следующих типов: электроды первого, второго рода, окислительно-восстановительные, мембранные электроды.
Электроды первого рода - это электроды, обратимые по катиону, общему с материлом электрода. Различают три разновидности электродов первого рода.
а) Металл М, погруженный в раствор соли того же металла. На поверхности таких электродов протекает обратимая реакция:
Мn+ + пе = М
Реальный потенциал такого электрода первого рода зависит от активности a(Mn+) катионов металла и описывается уравнениями (5)-(8).
В общем случае для любой температуры:
꞊E˚+ RTnFln a(Mn+)
Для комнатной температуры:
꞊E˚+ 0,059nln a(Mn+)
При малых концентрациях c(Mn+), когда активность a(Mn+)катионов металла приблизительно равна их концентрации:
꞊E˚+ RTnFln c(Mn+)
Для комнатной температуры:
б)Газовые электроды, например, водородный электрод, в том числе и стандартный водородный электрод. Потенциал обратимо работающего газового водородного электрода определяется активностью ионов водорода, т.е. величиной рН раствора, и при комнатной температуре равен:
꞊E˚+ 0,059 lg а(Н30+) = 0,059 lg а(Н3О+) = -0,059рН
поскольку для водородного электрода стандартный потенциал принимается равным нулю (£° =0), а в соответствии с электродной реакцией:
Н++е = Н
число электронов, участвующих в этой реакции, равно единице: п = 1. в)Амальгамные электроды, представляющие собой амальгаму металла, погруженную в раствор, содержащий катионы того же металла. Потенциал таких электродов первого рода зависит от активности a(Mn+)катионов металла в растворе и активности я(М) металла в амальгаме: ꞊E˚+ RTnFlna(Mn+)a(M)
Амальгамные электроды обладают высокой обратимостью. Электроды второго рода обратимы по аниону. Различают следующие виды электродов второго рода. а) Металл, поверхность которого покрыта малорастворимой солью этого же металла, погруженный в раствор, содержащий анионы, входящие в состав этой малорастворимой соли. Примером могут служить хлорсеребряный электрод Ag|AgCl, КС1 или каломельный электрод Hg|Hg2Cl2, КС1. Хлорсеребряный электрод состоит из серебряной проволоки, покрытой малорастворимой в воде солью AgCI, погруженной в водный раствор хлорида калия. На хлорсеребряном электроде протекает обратимая реакция Каломельный электрод состоит из металлической ртути, покрытой пастой малорастворимого хлорида ртути(1) Hg2Cl2 - каломели, контактирующей с водным раствором хлорида калия. На каломельном электроде протекает обратимая реакция: Cl2 + 2е = 2Hg + 2СГ.
Реальный потенциал электродов второго рода зависит от активности анионов и для обратимо работающего электрода, на котором протекает реакция: Ne = М + Аn- описывается уравнениями Нернста (9)-(12). В общем случае при любой приемлемой температуре Т: ꞊E˚- RTnFln a(An-)
Для комнатной температуры: ꞊E˚- 0,059nln a(An-)
Для условий, в которых активность анионов приблизительно равна их концентрации с(А"~):
E꞊E˚- RTnFln c(An-)
Для комнатной температуры: ꞊E˚- 0,059nln c(An-)
Так, например, реальные потенциалы Е1 и E2 соответственно хлор-серебряного и каломельного электродов при комнатной температуре можно представить в виде: ꞊E1˚- 0,0591g a(Cl-),꞊E2˚- 0,0591g a(Cl-).
Электроды второго рода обладают высокой обратимостью и стабильны в работе, поэтому их часто используют в качестве электродов сравнения, способных устойчиво поддерживать постоянное значение потенциала. б) Газовые электроды второго рода, например, хлорный электрод Pt, Cl2 КС1. Газовые электроды второго рода в количественном потенциометрическом анализе применяются редко. Окислительно-восстановительные электроды состоят из инертного материала (платина, золото, вольфрам, титан, графит и др.), погруженного в раствор, содержащий окисленную Ох и восстановленную Red формы данного вещества. Существуют две разновидности окислительно-восстановительных электродов: а)электроды, потенциал которых не зависит от активности ионов водорода, например, Pt | FeCl3, FeCI2, Pt | K3, K4 и т.д.; б)электроды, потенциал которых зависит от активности ионов водорода, например, хингидронный электрод. На окислительно-восстановительном электроде, потенциал которого не зависит от активности ионов водорода, протекает обратимая реакция:
Ох + пе = Red
Реальный потенциал такого окислительно-восстановительного электрода зависит от активности окисленной и восстановленной форм данного вещества и для обратимо работающего электрода описывается, в зависимости от условий (по аналогии с вышерассмотренными потенциалами), уравнениями Нернста (13)-(16): ꞊E˚+ RTnFln a (Ox)a (Red)꞊E˚+ 0,059nlg a (Ox)a (Red)꞊E˚+ RTnFln c(Ox)c (Red)꞊E˚+ 0,059nlg c (Ox)c(Red)
Если в электродной реакции участвуют ионы водорода, то их активность (концентрацию) учитывают в соответствующих уравнениях Нернста для каждого конкретного случая. Мембранные, или ион-селективные, электроды - электроды, обратимые по тем или иным ионам (катионам или анионам), сорбируемым твердой или жидкой мембраной. Реальный потенциал таких электродов зависит от активности тех ионов в растворе, которые сорбируются мембраной. Мембранные электроды с твердой мембраной содержат очень тонкую мембрану, по обе стороны которой находятся разные растворы, содержащие одни и те же определяемые ионы, но с неодинаковой концентрацией: раствор (стандартный) с точно известной концентрацией определяемых ионов и анализируемый раствор с неизвестной концентрацией определяемых ионов. Вследствие различной концентрации ионов в обоих растворах ионы на разных сторонах мембраны сорбируются в неодинаковых количествах, неодинаков и возникающий при сорбции ионов электрический заряд на разных сторонах мембраны. Как результат возникает мембранная разность потенциалов. Определение ионов с применением мембранных ион-селективных электродов называют ионометрией. Как уже говорилось выше, при потенциометрических измерениях электрохимическая ячейка включает два электрода - индикаторный электрод и электрод сравнения. Величина ЭДС, генерируемой в ячейке, равна разности потенциалов этих двух электродов. Поскольку потенциал электрода сравнения в условиях проведения потенциометрического определения остается постоянным, то ЭДС зависит только от потенциала индикаторного электрода, т.е. от активностей (концентраций) тех или иных ионов в растворе. На этом и основано потенциометрическое определение концентрации данного вещества в анализируемом растворе. Для потенциометрического определения концентрации вещества в растворе применяют как прямую потенциометрию, так и потенциометрическое титрование, хотя второй способ используется намного чаще первого.
Определение концентрации вещества в прямой потенциометрии проводят обычно методом градуировочного графика или методом добавок стандарта. а) Метод градуировочного графика. Готовят серию из 5-7 эталонных растворов с известным содержанием определяемого вещества. Концентрация определяемого вещества и ионная сила в эталонных растворах не должны сильно отличаться от концентрации и ионной силы анализируемого раствора: в этих условиях уменьшаются ошибки определения. Ионную силу всех растворов поддерживают постоянной.введением индифферентного электролита. Эталонные растворы последовательно вносят в электрохимическую (потенциометрическую) ячейку. Обычно эта ячейка представляет собой стеклянный химический стакан, в который помещают индикаторный электрод и электрод сравнения. Измеряют ЭДС эталонных растворов, тщательно промывая дистиллированной водой электроды и стакан перед заполнением ячейки каждым эталонным раствором. По полученным данным строят градуировочный график в координатах ЭДС-lg с, где с - концентрация определяемого вещества в эталонном растворе. Обычно такой график представляет собой прямую линию. Затем в электрохимическую ячейку вносят (после промывания ячейки дистиллированной водой) анализируемый раствор и измеряют ЭДС ячейки. По градуировочному графику находят lg с(Х), где с(Х) - концентрация определяемого вещества в анализируемом растворе. б) Метод добавок стандарта. В электрохимическую ячейку вносят известный объем V(X) анализируемого раствора с концентрацией с(Х) и измеряют ЭДС ячейки. Затем в тот же раствор прибавляют точно измеренный небольшой объем стандартного раствора V(ст) с известной, достаточно большой, концентрацией с(ст) определяемого вещества и снова определяют ЭДС ячейки. Рассчитывают концентрацию с(Х) определяемого вещества в анализируемом растворе по формуле (10.17):
с(Х)= с(ст) V (ст)V X+ V (ст)
где △E - разность двух измеренных значений ЭДС, п - число электронов, участвующих в электродной реакции.
Применение прямой потенциометрии. Метод применяется для определения концентрации ионов водорода (рН растворов), анионов, ионов металлов (ионометрия). Большую роль при использовании прямой потенциометрии играют выбор подходящего индикаторного электрода и точное измерение равновесного потенциала. При определении рН растворов в качестве индикаторных используют электроды, потенциал которых зависит от концентрации ионов водорода: стеклянный, водородный, хингидронный и некоторые другие. Чаще применяют мембранный стеклянный электрод, обратимый по ионам водорода. Потенциал такого стеклянного электрода определяется концентрацией ионов водорода, поэтому ЭДС цепи, включающей стеклянный электрод в качестве индикаторного, описывается при комнатной температуре уравнением: K + 0,059рН,
где постоянная К зависит от материала мембраны, природы электрода сравнения. Стеклянный электрод позволяет определять рН в интервале рН = 0-10 (чаще - в диапазоне рН = 2-10) и обладает высокой обратимостью и стабильностью в работе. Хингидронный электрод, часто применявшийся ранее, - это окислительно-восстановительный электрод, потенциал которого зависит от концентрации ионов водорода. Он представляет собой платиновую проволоку, погруженную в раствор кислоты (обычно НС1), насыщенный хингидроном - эквимолекулярным соединением хинона с гидрохиноном состава С6Н402 С6Н4(ОН)2 (темно-зеленый порошок, малорастворимый в воде). Схематическое обозначение хингидронного электрода: Pt | хингидрон, НС1.
На хингидронном электроде протекает окислительно-восстановительная реакция:
С6Н402 + 2Н+ + 2е = С6Н4(ОН)2
Потенциал хингидронного электрода при комнатной температуре описывается формулой E°-0,059рН.
Хингидронный электрод позволяет измерять рН растворов в интервале рН = 0-8,5. При рН < 0 хингидрон гидролитически расщепляется: при рН > 8,5 гидрохинон, являющийся слабой кислотой, вступает в реакцию нейтрализации, Хингидронный электрод нельзя применять в присутствии сильных окислителей и восстановителей. Мембранные ион-селективные электроды используют, как уже отмечалось выше, в ионометрии в качестве индикаторных для определения различных катионов (Li+, Na+, К+ Mg2t, Са2+, Cd2+, Fe2+, Ni2+ и др.) ианионов (F-, Сl-, Вг-,I-, S2- и др.). К достоинствам прямой потенциометрии относятся простота и быстрота проведения измерений, для измерений требуются небольшие объемы растворов.
3Потенциометрическое титрование
Потенциометрическое титрование - способ определения объема титранта, затраченного на титрование определяемого вещества в анализируемом растворе, путем измерения ЭДС (в процессе титрования) с помощью гальванической цепи, составленной из индикаторного электрода и электрода сравнения. При потенциометрическом титровании анализируемый раствор, находящийся в электрохимической ячейке, титруют подходящим титрантом, фиксируя конец титрования по резкому изменению ЭДС измеряемой цепи - потенциала индикаторного электрода, который зависит от концентрации соответствующих ионов и резко изменяется в точке эквивалентности. Измеряют изменение потенциала индикаторного электрода в процессе титрования в зависимости от объема прибавленного титранта. По полученным данным строят кривую потенциометрического титрования и по этой кривой определяют объем израсходованного титранта в ТЭ. При потенциометрическом титровании не требуется использование индикаторов, изменяющих окраску вблизи ТЭ. Применение потенциометрического титрования. Метод универсальный, его можно применять для индикации конца титрования во всех типах титрования: кислотно-основном, окислительно-восстановительном, комплексиметрическом, осадительном, при титровании в неводных сре-дах. В качестве индикаторных используют стеклянный, ртутный, ионселективные, платиновый, серебряный электроды, а в качестве электродов сравнения - каломельный, хлорсеребряный, стеклянный. Метод обладает высокой точностью, большой чувствительностью: позволяет проводить титрование в мутных, окрашенных, неводных средах, раздельно определять компоненты смеси в одном анализируемом растворе, например, раздельно определять хлорид- и иодид-ионы при аргентометрическом титровании. Методами потенциометрического титрования анализируют многие лекарственные вещества, например, аскорбиновую кислоту, сульфамидные препараты, барбитураты, алкалоиды и др. Основателем кондуктометрического анализа считается немецкий физик и физико-химик Ф.В.Г. Кольрауш (1840-1910), который впервые в 1885 г. предложил уравнение, устанавливающее связь между электропроводностью растворов сильных электролитов и их концентрацией. В середине 40-х гг. XX в. был разработан метод высокочастотного кондуктометрического титрования. С начала 60-х гг. XX в. стали использовать кондуктометрические детекторы в жидкостной хроматографии.
1 Принцип метода. Основные понятия
Кондуктометрический анализ (кондуктометрия) основан на использовании зависимости между электропроводностью (электрической проводимостью) растворов электролитов и их концентрацией. Об электропроводности растворов электролитов - проводников второго рода - судят на основании измерения их электрического сопротивления в электрохимической ячейке, которая представляет собой стеклянный сосуд (стакан) с двумя впаянными в него электродами, между которыми и находится испытуемый раствор электролита. Через ячейку пропускают переменный электрический ток. Электроды чаще всего изготовляют из металлической платины, которую для увеличения поверхности электродов покрывают слоем губчатой платины путем электрохимического осаждения из растворов платиновых соединений (электроды из платинированной платины). Во избежание осложнений,связанных с процессами электролиза и поляризации, кондуктометрические измерения проводят в переменном электрическом поле. Электрическое сопротивление R слоя раствора электролита между электродами, как и электрическое сопротивление проводников первого рода, прямо пропорционально длине (толщине) l этого слоя и обратно пропорционально площади S поверхности электродов:
R= ρ lS lkS
где коэффициент пропорциональности р называют удельным электрическим сопротивлением, а обратную величину к = 1/р - удельной электропроводностью (удельной электрической проводимостью). Так как электрическое сопротивление R измеряют в омах, а толщину l слоя раствора электролита - в см, площадь S поверхности электродов - в см2, то удельную электропроводность к измеряют в единицах Ом-1 см-1, или, поскольку Ом-1 - это сименс (См), то - в единицах См см-1.
По физическому смыслу удельная электропроводность - это электрическая проводимость слоя электролита, находящегося между сторонами куба с длиной сторон 1 см, численно равная току, проходящему через слой раствора электролита с площадью поперечного сечения 1 см2 при градиенте приложенного электрического потенциала 1 В/см. Удельная электропроводность зависит от природы электролита и растворителя, от концентрации раствора, от температуры. С увеличением концентрации раствора электролита его удельная электропроводность вначале возрастает, затем проходит через максимум, после чего уменьшается. Такой характер изменения удельной электропроводности обусловлен следующими причинами. Вначале с увеличением концентрации электролита возрастает число ионов - токпереносящих частиц - как для сильных, так и для слабых электролитов. Поэтому электропроводность раствора (проходящий через него электрический ток) повышается. Затем по мере роста концентрации раствора увеличиваются его вязкость (понижающая скорости движения ионов) и электростатические взаимодействия между ионами, что препятствует возрастанию электрического тока и при достаточно больших концентрациях способствует его уменьшению. В растворах слабых электролитов с ростом концентрации понижается степень диссоциации молекул электролита, что приводит к уменьшению числа ионов - токпроводящих частиц - и к понижению удельной электропроводности. В растворах сильных электролитов при высоких концентрациях возможно образование ионных ассоциатов (ионных двойников, тройников и т.п.), что также благоприятствует падению электропроводности. Удельная электропроводность растворов электролитов увеличивается с ростом температуры вследствие понижения вязкости растворов, что приводит к повышению скорости движения ионов, а для слабых электролитов - также и к увеличению степени их ионизации (диссоциации на ионы). Поэтому количественные кондуктометрические измерения необходимо проводить при постоянной температуре, термостатируя кондуктометрическую ячейку. Кроме удельной электропроводности в кондуктометрии используют эквивалентную электропроводность X и молярную электропроводность р. По физическому смыслу эквивалентная электропроводность X - это электрическая проводимость слоя раствора электролита толщиной 1 см, находящегося между одинаковыми электродами с такой площадью, чтобы объем раствора электролита, заключенного между ними, содержал 1 г-экв растворенного вещества. При этом за молярную массу эквивалента принимается молярная масса одинаковых частиц с единичным зарядовым числом («зарядом»), например,
Н+, Br - , 12Са2+, 13Fe3+ и т.д.
Эквивалентная электропроводность увеличивается с уменьшением концентрации раствора электролита. Максимальное значение эквивалентной электропроводности достигается при бесконечном разбавлении раствора. Эквивалентная электропроводность, как и удельная, возрастает с повышением температуры. Эквивалентная электропроводность X связана с удельной электропроводностью к соотношением (20): λ= 1000 kc
В прямой кондуктометрии концентрацию вещества в анализируемом растворе определяют по результатам измерений удельной электропроводности этого раствора. При обработке данных измерений используют два метода: расчетный метод и метод градуировочного графика. Расчетный метод. В соответствии с уравнением (10.20) молярная концентрация эквивалента с электролита в растворе может быть рассчитана, если известны удельная электропроводность к и эквивалентная электропроводность : c = 1000 kλ
Удельную электропроводность определяют экспериментально на основании измерения электрического сопротивления термостатированной кондуктометрической ячейки. Эквивалентная электропроводность раствора λ равна сумме подвижностей катиона λ+ и аниона Х λ -:
λ = λ + + λ-
Если подвижности катиона и аниона известны, то концентрацию можно рассчитать по формуле (24):
c = 1000 kλ + + λ-
Так поступают при определении методом прямой кондуктометрии концентрации малорастворимого электролита в его насыщенном растворе (сульфаты кальция, бария; галогениды серебра и др.). Метод градуировочного графика. Готовят серию эталонных растворов, каждый из которых содержит точно известную концентрацию определяемого вещества, измеряют их удельную электропроводность при постоянной температуре в термостатируемой кондуктометрической ячейке. По полученным данным строят градуировочный график, откладывая по оси абсцисс концентрацию эталонных растворов, а по оси ординат - значения удельной электропроводности. В соответствии с уравнением (24) построенный график в относительно небольшом диапазоне изменения концентраций обычно представляет собой прямую линию. В широком интервале изменения концентраций, когда подвижности катиона и аниона, входящие в уравнение (24), могут заметно изменяться, наблюдаются отклонения от линейной зависимости. Затем строго в тех же условиях измеряют удельную электропроводность к(Х) определяемого электролита в анализируемом растворе с неизвестной концентрацией с(Х) и по графику находят искомую величину с(Х). Так определяют, например, содержание бария в баритовой воде - насыщенном растворе гидроксида бария. Применение прямой кондуктометрии. Методу прямой кондуктометрии присущи простота, высокая чувствительность. Однако метод малоселективен. Прямая кондуктометрия имеет ограниченное применение в анализе. Она используется для определения растворимости малорастворимых электролитов, для контроля качества дистиллированной воды и жидких пищевых продуктов (молока, напитков и др.), для определения общего содержания солей в минеральной, морской, речной воде и в некоторых других случаях.
3 Кондуктометрическое титрование
При кондуктометрическом титровании за ходом титрования следят по изменению электропроводности анализируемого раствора, находящегося в кондуктометрической ячейке между двумя инертными электродами (обычно из платинированной платины). По полученным данным вычерчивают кривую кондуктометрического титрования, отражающую зависимость электропроводности титруемого раствора от объема прибавленного титранта. Конечную точку титрования находят чаще всего экстраполяцией участков кривой титрования в области изменения ее наклона.При этом не требуется применение индикаторов, изменяющих окраску вблизи ТЭ. В кондуктометрическом титровании используют различные типы реакций: кислотно-основные, окислительно-восстановительные, осадительные, процессы комплексообразования. Применение кондуктометрического титрования. Метод кондуктометрического титрования обладает рядом достоинств. Титрование можно проводить в мутных, окрашенных, непрозрачных средах. Чувствительность метода довольно высокая - до ~10~* моль/л; ошибка определения составляет от 0,1 до 2%. Анализ можно автоматизировать. К недостаткам метода относится малая селективность. Понятие о высокочастотном (радиочастотном) кондуктометрическом титровании. За ходом титрования следят с помощью модифицированной переменно-токовой кондуктометрической техники, в которой частота переменного тока может достигать порядка миллиона колебаний в секунду. Обычно электроды помещают (накладывают) на внешней стороне сосуда (кондуктометрической ячейки) для титрования, так что они не соприкасаются с титруемым раствором. По результатам измерений вычерчивают кривую кондуктометрического титрования. Конечную точку титрования находят экстраполяцией участков кривой титрования в области изменения ее наклона.
ГЛАВА 4. КОНДУКТОМЕТРИЧЕСКИЙ АНАЛИЗ (КОНДУКТОМЕТРИЯ)
4.1 Сущность метода
Полярографический анализ (полярография) основан на использовании следующих зависимостей между электрическими параметрами электрохимической (в данном случае - полярографической) ячейки, к которой прилагается внешний потенциал, и свойствами содержащегося в ней анализируемого раствора. а)В качественном полярографическом анализе используют связь между величиной приложенного на микроэлектроде внешнего электрического потенциала, при котором наблюдается восстановление (или окисление) анализируемого вещества на микроэлектроде в данных условиях, и природой восстанавливающегося (или окисляющегося) вещества. б)В количественном полярографическом анализе используют связь между величиной диффузионного электрического тока, и концентрацией определяемого (восстанавливающегося или окисляющегося) вещества в анализируемом растворе. Электрические параметры - величину приложенного электрического потенциала и величину Диффузионного тока - определяют при анализе получаемых поляризационных, или вольт-амперных, кривых, отражающих графически зависимость электрического тока в полярографической ячейке от величины приложенного потенциала микроэлектрода. Поэтому полярографию иногда называют прямой вольтамперометрией. Классический полярографический метод анализа с применением ртутного капающего (капельного) электрода был разработан и предложен в 1922 г. чешским ученым Ярославом Гейровским (1890-1967), хотя сам ртутный капающий электрод применялся чешским физиком Б. Кучерой еще в 1903 г. В 1925 г. Я. Гейровский и М. Шиката сконструировали первый полярограф, позволивший автоматически регистрировать поляризационные кривые. В дальнейшем были разработаны различные модификации полярографического метода. Величина среднего диффузионного тока iD определяется уравнением Ильковича (25): где К- коэффициент пропорциональности, с - концентрация (ммоль/л) полярографически активного вещества-деполяризатора; iD измеряют в микроамперах как разность между предельным током и остаточным током. Коэффициент пропорциональности К в уравнении Ильковича зависит от целого ряда параметров и равен
K=607nD12m23τ16
где п - число электронов, принимающих участие в электродной окислительно-восстановительной реакции; D - коэффициент диффузии восстанавливающегося вещества (см2/с); т - масса ртути, вытекающей из капилляра в секунду (мг); т - время образования (в секундах) капли ртути при потенциале полуволны (обычно оно составляет 3-5 с). Так как коэффициент диффузии D зависит от температуры, то и коэффициент пропорциональности К в уравнении Ильковича изменяется при изменении температуры. Для водных растворов в температурном интервале 20-50 °С коэффициент диффузии полярографичски активных веществ-деполяризаторов увеличивается примерно на 3% при росте температуры на один градус, что и приводит к повышению среднего диффузионного тока iD на ~1-2%. Поэтому полярографирование проводят при постоянной температуре, термостатируя полярографическую ячейку обычно при 25 ± 0,5 °С. Масса ртути т и время каплеобразования т зависят от характеристик ртутного капающего электрода и высоты столбика ртути в капилляре и в резервуаре, связанном с капилляром. Стеклянный капилляр ртутного капающего микроэлектрода обычно имеет внешний диаметр 3-7 мм, внутренний - от 0,03 до 0,05 мм, длину 6-15 см. Высота ртутного столбика от нижнего конца капилляра до верхнего уровня поверхности ртути в резервуаре составляет 40-80 см; Содержание индифферентного электролита в анализируемом полярографируемом растворе должно примерно в 100 раз превышать содержание определяемого вещества-деполяризатора, причем ионы фонового электролита не должны разряжаться в условиях проведения полярографирования до разряда полярографически активного вещества. Полярографирование проводят с использованием в качестве растворителя воды, водно-органических смесей (вода - этанол, вода - ацетон, вода - диметилформамид и др.) и неводных сред (этанол, ацетон, диметилформамид, диметилсульфоксид и т.д.). До начала полярографирования через анализируемый раствор пропускают ток инертного газа (азота, аргона и др.) для удаления растворенного кислорода, который также дает полярографическую волну вследствие восстановления по схеме:
2Н+ + 2е = Н202 Н202 + 2Н+ + 2е = 2Н20
Иногда - в случае щелочных растворов - вместо пропускания тока инертного газа в анализируемый раствор прибавляют небольшое количество активного восстановителя - сульфита натрия, метола, которые связывают растворенный кислород, реагируя с ним.
4.2 Количественный полярографический анализ
Из изложенного выше следует, что количественный полярографический анализ основан на измерении диффузионного тока iD как функции концентрации определяемого полярографически активного вещества- деполяризатора в полярографируемом растворе. При анализе получаемых полярограмм концентрацию определяемого вещества находят методами градуировочного графика, добавок стандарта, стандартных растворов. а)Метод градуировочного графика используют чаще всего. По этому методу готовят серию стандартных растворов, каждый из которых содержит точно известную концентрацию с определяемого вещества. Проводят полярографирование каждого раствора (после продувания через него тока инертного газа) в одинаковых условиях, получают полярограммы и находят значения Е12 (одинаковые для всех растворов) и диффузионного тока iD (разные для всех растворов). По полученным данным строят градуировочный график в координатах iD-c, представляющий собой обычно прямую линию в соответствии с уравнением Ильковича. Затем проводят полярографирование анализируемого раствора с неизвестной концентрацией с(Х) определяемого вещества, получают полярограмму, измеряют величину диффузионного тока iD (Х) и по градуировочному графику находят концентрацию с(Х). б)Метод добавок стандарта. Получают полярограмму анализируемого раствора с неизвестной концентрацией с(Х) определяемого вещества и находят величину диффузионного тока, т.е. высоту h полярограммы. Затем к анализируемому раствору прибавляют точно известное количество определяемого вещества, повышающее его концентрацию на величину c(st), снова проводят полярографирование и находят новое значение диффузионного тока - высоту полярограммы h + △h.
В соответствии с уравнением Ильковича (25) можно написать: h = Kc(X),△h = K c(st),
откуда
h△h = с(Х)c(st) и с(Х) = h△hc(st)
в)Метод стандартных растворов. В одинаковых условиях проводят полярографирование двух растворов: анализируемого раствора с неизвестной концентрацией с(Х) и стандартного раствора с точно известной концентрацией c(st) определяемого вещества. На полученных полярограммах находят высоты полярографических волн h(Х) и h(st), отвечающие диффузионному току при концентрациях соответственно с(Х) и c(st). Согласно уравнению Ильковича (25) имеем: (Х) = Кс(Х), h(st) = Kc(st),
Стандартный раствор готовят так, чтобы его концентрация была бы как можно ближе к концентрации определяемого раствора. При этом условии ошибка определения минимизируется.
3 Применение полярографии
Применение метода. Полярография используется для определения малых количеств неорганических и органических веществ. Разработаны тысячи методик количественного полярографического анализа. Предложены способы полярографического определения практически всех катионов металлов, ряда анионов (бромат-, иодат-, нитрат-, перманганат-ионов), органических соединений различных классов, содержащих диазогруппы, карбонильные, пероксидные, эпоксидные группы, двойные углерод-углеродные связи, а также связи углерод-галоген, азот-кислород, сера-сера. Метод - фармакопейный, применяется для определения салициловой кислоты, норсульфазола, витамина Вь алкалоидов, фолиевой кислоты, келлина в порошке и в таблетках, никотинамида, пиридоксина гидрохлорида, препаратов мышьяка, гликозидов сердечного действия, а также кислорода и различных примесей в фармацевтических препаратах. Метод обладает высокой чувствительностью (до 10"5-10Т6 моль/л); селективностью; сравнительно хорошей воспроизводимостью результатов (до ~2%); широким диапазоном применения; позволяет анализировать смеси веществ без их разделения, окрашенные растворы, небольшие объемы растворов (объем полярографической ячейки может составлять всего 1 мл); вести анализ в потоке раствора; автоматизировать проведение анализа." К недостаткам метода относятся токсичность ртути, ее довольно легкая окисляемость в присутствии веществ-окислителей, относительная сложность используемой аппаратуры. Другие варианты полярографического метода. Помимо описанной выше классической полярографии, использующей капающий ртутный микроэлектрод с равномерно возрастающим на нем электрическим потенциалом при постоянном электрическом токе, разработаны другие варианты полярографического метода - производная, дифференциальная, импульсная, осциллографическая полярография; переменно-токовая полярография - также в разных вариантах.
ГЛАВА 5. АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Сущность метода. Амперометрическое титрование (потенцио-статическое поляризационное титрование) - разновидность вольтамперометрического метода (наряду с полярографией). Оно основано на измерении величины тока между электродами электрохимической ячейки, к которым приложено некоторое напряжение, как функции объема прибавленного титранта. В соответствии с уравнением Ильковича (25): диффузионный ток iD в полярографической ячейке тем больше, чем выше концентрация с полярографически активного вещества. Если при прибавлении титранта в анализируемый титруемый раствор, находящийся в полярографической ячейке, концентрация такого вещества уменьшается или увеличивается, то соответственно падает или возрастает и диффузионный ток. Точку эквивалентности фиксируют по резкому изменению падения или роста диффузионного тока, что отвечает окончанию реакциит титруемого вещества с титрантом. Различают амперометрическое титрование с одним поляризуемым электродом, называемое также титрованием по предельному току, полярографическим или поляриметрическим титрованием, и амперометрическое титрование с двумя одинаковыми поляризуемыми электродами, или титрование «до полного прекращения тока», биамперометрическое титрование. Амперометрическое титрование с одним поляризуемым электродом. Оно основано на измерении тока в полярографической ячейке в зависимости от количества прибавленного титранта при постоянном внешнем потенциале на микроэлектроде, несколько превышающем потенциал полуволны на вольт-амперной кривой титруемого вещества X или титранта Т. Обычно выбранный внешний потенциал соответствует области предельного тока на полярограмме X или Т. Титрование ведут на установке, состоящей из источника постоянного тока с регулируемым напряжением, к которому последовательно присоединены гальванометр и полярографическая ячейка для титрования. Рабочим (индикаторным) электродом ячейки может служить ртутный капающий электрод, неподвижный или вращающийся платиновый либо графитовый электрод. При использовании твердых электродов необходимо перемешивание раствора во время титрования. В качестве электрода сравнения применяют хлор-серебряный или каломельный электроды. Фоном служат, в зависимости от условий, различные полярографически неактивные при данном потенциале электролиты (HN03, H2S04, NH4NO3 и др.). Вначале получают вольт-амперные кривые (полярограммы) для X и Т в тех же условиях, в которых предполагается проведение амперометрического титрования. На основании рассмотрения этих кривых выбирают значение потенциала, при котором достигается величина предельного тока полярографически активных X или Т. Выбранное значение потенциала поддерживают постоянным в течение всего процесса титрования. Используемая для амперометрического титрования концентрация титранта Т должна примерно в 10 раз превышать концентрацию X; при этом практически не требуется вводить поправку на разбавление раствора во время титрования. В остальном соблюдают все те условия, которые требуются для получения полярограмм. Требования к термостатированию - менее строгие, чем при прямом полярографировании, поскольку конец титрования определяется не по абсолютному значению диффузионного тока, а по резкому изменению его величины. В полярографическую ячейку вносят анализируемый раствор, содержащий X, и прибавляют небольшими порциями титрант Т, измеряя каждый раз ток i. Величина тока i зависит от концентрации полярографически активного вещества. В точке эквивалентности величина i резко изменяется. По результатам амперометрического титрования строят кривые титрования. Кривая амперометрического титрования - это графическое представление изменения величины тока / в зависимости от объема V прибавленного титранта. Кривая титрования строится в координатах ток i - объем V прибавленного титранта Т (или степень оттитрованности). В зависимости от природы титруемого вещества X и титранта Т кривые амперометрического титрования могут быть различного типа. Биамперометрическое титрование ведут при энергичном перемеши-вании раствора на установке, состоящей из источника постоянного тока с потенциометром, с которого регулируемая разность потенциалов (0,05- 0,25 В) подается через чувствительный микроамперметр на электроды электрохимической ячейки. В последнюю перед проведением титрования вносят титруемый раствор и прибавляют порциями титрант до резкого прекращения или появления тока, о чем судят по показанию микроамперметра. Используемые в электрохимической ячейке платиновые электроды периодически очищают, погружая их на ~30 минут в кипящую концентрированную азотную кислоту, содержащую добавки хлористого железа, с последующим промыванием электродов водой. Биамперометрическое титрование - фармакопейный метод; применяется в иодометрии, нитритометрии, акваметрии, при титровании в не водных средах.
ГЛАВА 6. КУЛОНОМЕТРИЧЕСКИЙ АНАЛИЗ (КУЛОНОМЕТРИЯ)
1 Принципы метода электрохимический кондуктометрия титрование кулонометрия Кулонометрический анализ (кулонометрия) основан на использовании зависимости между массой т вещества, прореагировавшего при электролизе в электрохимической ячейке, и количеством электричества Q, прошедшего через электрохимическую ячейку при электролизе только этого вещества. В соответствии с объединенным законом электролиза М Фарадея масса т (в граммах) связана с количеством электричества Q (в кулонах) соотношением (27) где М - молярная масса вещества, прореагировавшего при электролизе, г/моль; п - число электронов, участвующих в электродной реакции; 96487 Кл/моль - число Фарадея.
Количество электричества Q (в Кл), прошедшее при электролизе через электрохимическую ячейку, равно произведению электрического тока i (в А) на время электролиза τ (в с):
Если измерено количество электричества Q, то согласно (27) можно рассчитать массу т. Это справедливо в том случае, когда все количество электричества Q, прошедшее при электролизе через электрохимическую ячейку, израсходовано только на электролиз данного вещества; побочные процессы должны быть исключены. Другими словами, выход (эффективность) по току должен быть равен 100%. Поскольку в соответствии с объединенным законом электролиза М. Фарадея для определения массы т (г) прореагировавшего при электролизе вещества необходимо измерить количество электричества Q, затраченное на электрохимическое превращение определяемого вещества, в кулонах, то метод и назван кулонометрией. Главная задача кулонометрических измерений - как можно более точно определить количество электричества Q. Кулонометрический анализ проводят либо в амперостатическом (гальваностатическом) режиме, т.е. при постоянном электрическом токе i=const, либо при контролируемом постоянном потенциале рабочего электрода (потенциостатическая кулонометрия), когда электрический ток изменяется (уменьшается) в процессе электролиза. В первом случае для определения количества электричества Q достаточно как можно более точно измерить время электролиза т(с), постоянный ток /(А) и рассчитать величину Q по формуле (10.28). Во втором случае величину Q определяют либо расчетным способом, либо с помощью химических кулонометров. Различают прямую кулонометрию и косвенную кулонометрию (кулонометрическое титрование).
Сущность метода. Прямую кулонометрию при постоянном токе применяют редко. Чаще используют кулонометрию при контролируемом постоянном потенциале рабочего электрода или прямую потенциостатическую кулонометрию. В прямой потенциостатической кулонометрии электролизу подвергают непосредственно определяемое вещество. Измеряют количество электричества, затраченное на электролиз этого вещества, и по уравнению рассчитывают массу т определяемого вещества. В процессе электролиза потенциал рабочего электрода поддерживают постоянным, Е=const, для чего обычно используют приборы - потенциостаты. Постоянное значение потенциала Е выбирают предварительно на основании рассмотрения вольт-амперной (поляризационной) кривой, построенной в координатах ток i - потенциал Е (как это делают в полярографии), полученной в тех же условиях, в которых будет проводиться электролиз. Обычно выбирают значение потенциала Е, соответствующее области предельного тока для определяемого вещества и несколько превышающее его потенциал полуволны Е12 (на -0,05-0,2 В). При этом значении потенциала, как и в полярографии, фоновый электролит не должен подвергаться электролизу. По мере протекания процесса электролиза при постоянном потенциале электрический ток в ячейке уменьшается, так как понижается концентрация электроактивного вещества, участвующего в электродной реакции. При этом электрический ток уменьшается со временем по экспо-ненциальному закону от начального значения i0 в момент времени т = О до значения i в момент времени т:
где коэффициент к зависит от природы реакции, геометрии электрохимической ячейки, площади рабочего электрода, коэффициента диффузии определяемого вещества, скорости перемешивания раствора и его объема. Способы определения количества электричества, прошедшего через раствор, в прямой потепциостатической кулонометрии. Величину Q можно определить расчетными способами либо с помощью химического кулонометра. а)Расчет вечичины Q по площади под кривой зависимости i от т. Для определения Q без заметной ошибки способ требует практически полного завершения процесса электролиза, т.е. длительного времени. На практике, как уже отмечалось выше, измеряют площадь при значении т, соответствующем 0,001i0 (0,1% от i0).
б)Расчет величины Q на основе зависимости In / от т. В соответствии имеем:
Q = 0∞i0e-kτdτ=i00∞e-kτdτ=i0k
Поскольку
∞i0e-kτdτ= - k-1 e-k∞-e-k0= k-10-1=k-1
Применение прямой кулонометрии. Метод обладает высокими селективностью, чувствительностью (до 10~8-10~9 г или до ~10~5 моль/л), воспроизводимостью (до ~1-2%), позволяет определять содержание микропримесей. К недостаткам метода относятся большие трудоемкость и длительность проведения анализа, необходимость наличия дорогостоящей аппаратуры. Прямую кулонометрию можно применять для определения - при катодном восстановлении - ионов металлов, органических нитро- и галогенпроизводных; при анодном окислении - хлорид-, бромид-, иодид-,тиоцианат-анионы, ионы металлов в низших степенях окисления при переводе их в более высокие состояния окисления, например: As(IH) -> As(V),Cr(II) -> Cr(III), Fe(II) -» Fe(III), T1(I) -> Tl(III) и т.д. В фармацевтическом анализе прямую кулонометрию применяют для определения аскорбиновой и пикриновой кислот, новокаина, оксихинолина и в некоторых других случаях. Как отмечалось выше, прямая кулонометрия довольно трудоемка и продолжительна. Кроме того, в ряде случаев начинают заметно протекать побочные процессы еще до завершения основной электрохимической реакции, что снижает выход по току и может привести к значительным ошибкам анализа. Поэтому чаще применяют косвенную кулонометрию - кулонометрическое титрование.
3 Кулонометрическое титрование
Сущность метода. При кулонометрическом титровании определяемое вещество X, находящееся в растворе в электрохимической ячейке, реагирует с «титрантом» Т - веществом, непрерывно образующемся (генерируемом) на генераторном электроде при электролизе вспомогательного вещества, также присутствующего в растворе. Окончание титрования - момент, когда все определяемое вещество X полностью прореагирует с генерируемым «титрантом» Т, фиксируют либо визуально индикаторным методом, вводя в раствор соответствующий индикатор, меняющий окраску вблизи ТЭ, либо с помощью инструментальных методов - потенциометрически, амперометрически, фотометрически. Таким образом, при кулонометрическом титровании титрант не прибавляется из бюретки в титруемый раствор. Роль титранта играет вещество Т, непрерывно генерируемое при электродной реакции на генераторном электроде. Очевидно, имеется аналогия между обычным титрованием, когда титрант вводится извне в титруемый раствор и по мере его прибавления реагирует с определяемым веществом, и генерацией вещества Т, которое по мере своего образования также реагирует с определяемым веществом. Поэтому рассматриваемый метод и получил название «кулонометрическое титрование». Кулонометрическое титрование проводят в амперостатическом (гальваностатическом) или в потенциостатическом режиме. Чаще кулонометрическое титрование проводят в амперостатическом режиме, поддерживая электрический ток постоянным в течение всего времени электролиза. Вместо объема прибавленного титранта в кулонометрическом титровании измеряют время т и ток i электролиза. Процесс образования вещества Т в кулонометрической ячейке во время электролиза называется генерация титранта. Кулонометрическое титрование при постоянном токе. При кулонометрическом титровании в амперостатическом режиме (при посто-янном токе) измеряют время т, в течение которого проводился электролиз, и количество электричества Q, израсходованное при электролизе, рассчитывают по формуле, после чего находят массу определяемого вещества X по соотношению. Так, например, стандартизацию раствора хлороводородной кислоты НС1 методом кулонометрического титрования проводят путем титрования ионов водорода Н30+ стандартизуемого раствора, содержащего НС1, электрогенерируемыми на платиновом катоде гидроксид-ионами ОН- при электролизе воды:
Н20 + 2е = 20Н- + Н2
Образовавшийся титрант - гидроксид-ионы - реагирует с ионамиН30+ в растворе:
Н30+ + ОН- = 2Н20 Титрование ведут в присутствии индикатора фенолфталеина и прекращают при появлении светло-розовой окраски раствора. Зная величину постоянного тока i (в амперах) и время т (в секундах), затраченное на титрование, рассчитывают по формуле (28) количество электричества Q (в кулонах) и по формуле (27) - массу (в граммах) прореагировавшей НС1, содержавшуюся в аликвоте стандартизуемого раствора НС1, внесенного в кулонометрическую ячейку (в генераторный сосуд). Условия проведения кулонометрического титрования. Из вышеизложенного следует, что условия проведения кулонометрического титрования должны обеспечить 100%-ный выход по току. Для этого необходимо выполнять, по крайней мере, следующие требования. а)Вспомогательный реагент, из которого на рабочем электроде гнерируется титрант, должен присутствовать в растворе в большом избытке по отношению к определяемому веществу (~ 1000-кратный избыток). В этих условиях обычно устраняются побочные электрохимические реакции, основная из которых - это окисление или восстановление фонового электролита, например, ионов водорода:
Н+ + 2е = Н2
б)Величина постоянного тока i=const при проведении электролиза должна быть меньше величины диффузионного тока вспомогательного реагента во избежание протекания реакции с участием ионов фонового электролита. в)Необходимо как можно точнее определять количество электричества, израсходованное при проведении электролиза, для чего требуется точно фиксировать начало и конец отсчета времени и величину тока электролиза. Кулонометрическое титрование при постоянном потенциале. Потенциостатический режим в кулонометрическом титровании используется реже. Кулонометрическое титрование в потенциостатическом режиме ведут при постоянном значении потенциала, соответствующем потенциалу разряда вещества на рабочем электроде, например, при катодном восстановлении катионов металлов М"* на платиновом рабочем электроде. По мере протекания реакции потенциал остается постоянным до тех пор, пока прореагируют все катионы металла, после чего он резко уменьшается, поскольку в растворе уже нет потенциалопределяющих катионов металла. Применение кулонометрического титрования. В кулонометрическом титровании можно использовать все типы реакций титриметрического анализа: кислотно-основные, окислительно-восстановительные, осадительные, реакции комплексообразования. Так, малые количества кислот можно определять кулонометрическим кислотно-основным титрованием электрогенерированными ОН--ионами, образующимися при электролизе воды на катоде:
Н20 + 2е = 20Н" + Н2
Можно титровать и основания ионами водорода Н+, генерируемыми на аноде при электролизе воды:
Н20-4е = 4Н+ + 02
При окислительно-восстановительном бромометрическом кулонометрическом титровании можно определять соединения мышьяка(Ш), сурьмы(Ш), иодиды, гидразин, фенолы и другие органические вещества. В роли титранта выступает электрогенерируемый на аноде бром:
ВГ -2е = Вг2
Осадительным кулонометрическим титрованием можно определять галогенид-ионы и органические серосодержащие соединения электрогенерированными катионами серебра Ag+, катионы цинка Zn2+ - электрогенерированными ферроцианид-ионами и т.д. Комплексонометрическое кулонометрическое титрование катионов металлов можно проводить анионами ЭДТА, электрогенерированными на катоде из комплексоната ртути(И). Кулонометрическое титрование обладает высокой точностью, широким диапазоном применения в количественном анализе, позволяет определять малые количества веществ, малостойкие соединения (посколькуони вступают в реакции сразу же после их образования), например,меди(1), серебра(Н), олова(П), титана(Ш), марганца(Ш), хлора, брома и др. К достоинствам метода относится также и то, что не требуются приготовление, стандартизация и хранение титранта, так как он непрерывно образуется при электролизе и сразу же расходуется в реакции с определяемым веществом. ЗАКЛЮЧЕНИЕ
Электрохимические методы анализа основаны на процессах, протекающих на электродах или межэлектродном пространстве. Электрохимические методы анализа являются одними из старейших физико-химических методов анализа (некоторые описаны в конце 19 в.). Их достоинством является высокая точность и сравнительная простота, как оборудования, так и методики анализа. Высокая точность определяется весьма точными закономерностями, используемыми в электрохимических методах анализа, например, закон Фарадея. Большим удобством является то, что в них используют электрические воздействия, и то, что результат этого воздействия (отклик) тое получается в виде электрического сигнала. Это обеспечивает высокую скорость и точность отсчета, открывает широкие возможности для автоматизации. Электрохимические методы анализа отличаются хорошей чувствительностью и селективностью, в ряде случаев их можно отнести к микроанализу, так как для анализа иногда достаточно менее 1 мл раствора. Инструментом их служит электрохимическая ячейка, представляющая собой сосуд с раствором электролита, в который погружены как минимум два электрода. В зависимости от решаемой задачи различными могут быть форма и материал сосуда, число и природа электродов, раствора, условия анализа (прилагаемое напряжение (ток) и регистрируемый аналитический сигнал, температура, перемешивание, продувка инертным газом и тому подобное). Определяемое вещество может входить как в состав электролита, заполняющего ячейку, так и в состав одного из электродов. Электрохимические методы анализа играют большую роль в современном мире. В наше время особенно важна забота об экологии. С помощью этих методов можно определить содержание огромного количества различных органических и неорганических веществ. Сейчас они более эффективны для определения опасных веществ.
Электрохимические методы анализа - это совокупность методов качественного и количественного анализа, основанных на электрохимических явлениях, происходящих в исследуемой среде или на границе раздела фаз и связанных с изменением структуры, химического состава или концентрации анализируемого вещества.
Электpохимические методы анализа (ЭХМА) основаны на процессах, пpотекающих на электpодах или межэлектpодном пpостpанстве. Их достоинством является высокая точность и сpавнительная пpостота как обоpудования, так и методик анализа. Высокая точность опpеделяется весьма точными закономеpностями используемыми в ЭХМА. Большим удобством является то, что в этом методе используют электpические воздействия, и то, что pезультат этого воздействия (отклик) тоже получается в виде электрического сигнала. Это обеспечивает высокую скоpость и точность отсчета, откpывает шиpокие возможности для автоматизации. ЭХМА отличаются хорошей чувствительностью и селективностью, в pяде случаев их можно отнести к микpоанализу, так как для анализа иногда достаточно менее 1 мл pаствоpа.
По разновидностям аналитического сигнала подразделяют на:
1) кондуктометрию - измерение электропроводности исследуемого раствора;
2) потенциометрию - измерение бестокового равновесного потенциала индикаторного электрода, для которого исследуемое вещество является потенциоопределяющим;
3) кулонометрию - измерение количества электричества, необходимого для полного превращения (окисления или восстановления) исследуемого вещества;
4) вольтамперометрию - измерение стационарных или нестационарных поляризационных характеристик электродов в реакциях с участием исследуемого вещества;
5) электрогравиметрию - измерение массы вещества, выделенного из раствора при электролизе.
27. Потенциометрический метод.
потенциометрию - измерение бестокового равновесного потенциала индикаторного электрода, для которого исследуемое вещество является потенциоопределяющим.
А) стандартная(электрод сравнения) – имеет постоянный потенциал, не зависящий от внеш. Условий
Б) индивидуальный электрод – его потенциал зависит от концентрации вещества.
Потенциал зависит от концентрации: Е = f(c)
Уравнение Нериста Е= Е° + lna kat
E ° - стандарт. Электрон. Потенциал (const )
R – универ. Газовая постоянная const )
Т – абсолютная темп (t )- +273 °
.п – число электронов участвующ. В окис./восст. Реакции
. а – активная концентрация
Метод потенциометрии
Ионометрия потенциометрирование (к исслед. Р-ру небольш. Порциями добавляется стандарт.р-р(титран), после каждого прибавления измеряют потенциал.- Е)
Точка эквивалентности
ЕСх Vх = l т *Vт
28. Кондуктометрический метод.
кондуктометрия- измерение электропроводности исследуемого раствора.
Кондуктометрическое титрование
Кондуктометр (прибор)
Кондуктометрический анализ (кондуктометрия) основан на использовании зависимости между электропроводностью (электрической проводимостью) растворов электролитов и их концентрацией.
Об электропроводности растворов электролитов - проводников второго рода - судят на основании измерения их электрического сопротивления в электрохимической ячейке, которая представляет собой стеклянный сосуд (стакан) с двумя впаянными в него электродами, между которыми и находится испытуемый раствор электролита. Через ячейку пропускают переменный электрический ток. Электроды чаще всего изготовляют из металлической платины, которую для увеличения поверхности электродов покрывают слоем губчатой платины путем электрохимического осаждения из растворов платиновых соединений (электроды из платинированной платины).
29.Полярография.
Полярография - метод качественного и количественного химического анализа, основанный на получении кривых зависимости величины тока от напряжения в цепи состоящей из исследуемого раствора и погруженных в него электродов, один из которых сильно поляризующийся, а другой практически неполяризующийся. Получение таких кривых - полярограмм - производят при помощи полярографов.
Полярографический метод характеризуется большой чувствительностью. Для выполнения анализа обычно достаточно 3-5 мл исследуемого раствора. Анализ при помощи авторегистрирующего полярографа длится всего около 10 минут. Полярографию используют для определения в объектах биологического происхождения содержания ядовитых веществ (например, соединений ртути, свинца, таллия и др.), для определения степени насыщения крови кислородом, исследования состава выдыхаемого воздуха, вредных веществ в воздухе промышленных предприятий.Полярографический метод анализа обладает большой чувствительностью и дает возможность определять вещества при очень незначительной (до 0,0001%) концентрации их в растворе.
30.Классификация спектральных методов анализа. Понятие спектра.
Спектральный анализ – это совокупность методов определения кач.и колич. Состава, а так же структуры вещества (основанных на взаимодействии исслед.объекта с различными типами излучения.)
Все спектроскопические методы основаны на взаимодействии атомов, молекул или ионов, входящих в состав анализируемого вещества, с электромагнитным излучением. Это взаимодействие проявляется в поглощении или испускании фотонов (квантов). В зависимости от характера взаимодействия пробы с электромагнитным излучением выделяют две группы методов –
Эмиссионные и абсорбционные. В зависимости от того, какие частицы формируют аналитический сигнал, различают методы атомной спектроскопии и методы молекулярной спектроскопии
Эмиссионная
В эмиссионных методах анализируемая проба в результате ее возбуждения излучает фотоны.
абсорбционная
В абсорбционных методах излучение постороннего источника пропускают через пробу, при этом часть квантов избирательно поглощается атомами или молекулами
Спектр - распределение значений физической величины (обычно энергии, частоты или массы). Графическое представление такого распределения называется спектральной диаграммой. Обычно под спектром подразумевается электромагнитный спектр - спектр частот (или то же самое, что энергий квантов) электромагнитного излучения.
1.отражение света
2.поворот пучка света(дефракция)
3.рассеивание света: нефелометрия,турбидиметрия
4.поглощение света
5переизлучение
А)фосфоресценция (длится долго)
Б)флуоресценция(очень короткая)
По характеру распределения значений физической величины спектры могут быть дискретными (линейчатыми), непрерывными (сплошными), а также представлять комбинацию (наложение) дискретных и непрерывных спектров.
Примерами линейчатых спектров могут служить масс-спектры и спектры связанно-связанных электронных переходов атома; примерами непрерывных спектров - спектр электромагнитного излучения нагретого твердого тела и спектр свободно-свободных электронных переходов атома; примерами комбинированных спектров - спектры излучения звёзд, где на сплошной спектр фотосферы накладываются хромосферные линии поглощения или большинство звуковых спектров.
31.Фотометрия: принцип метода, применение в суд.исследованиях.
Фотометрия – спектральный метод основан на поглощении электромагнитного излучения видимого и ближнего ультрафиолетового диапазона (метод основан на поглощении света)
Молекулярная Атомная
Спектроскопия спектроскопия(В электрон.Анализе)
Кювета – через нее пропускают свет
l
I (интенсивность выход.света)
I° – интенсивность падающего света.
Фотометрия – раздел физической оптики и измерительной техники, посвященный методам исследования энергетических характеристик оптического излучения в процессе его испускания, распространения в различных средах и взаимодействия с телами. Фотометрию проводят в диапазонах инфракрасного (длины волн – 10 –3…7 10 –7 м), видимого (7 10 –7…4 10 –7 м) и ультрафиолетового (4 10 –7…10 –8 м) оптических излучений. При распространении электромагнитного излучения оптического диапазона в биологической среде наблюдаются ряд основных эффектов: поглощение и рассеивание излучения атомами и молекулами среды, рассеивание на частицах неоднородностей среды, деполяризация излучения. Регистрируя данные взаимодействия оптического излучения со средой, можно определить количественные параметры, связанные с медико-биологическими характеристиками исследуемого объекта. Для измерения фотометрических величин применяют приборы – фотометры. С точки зрения фотометрии, свет – это излучение, способное вызывать ощущение яркости при воздействии на человеческий глаз. В основе фотометрии как науки лежит разработанная А. Гершуном теория светового поля.
Существуют два общих метода фотометрии: 1) визуальная фотометрия, в которой при выравнивании механическими или оптическими средствами яркости двух полей сравнения используется способность человеческого глаза ощущать различия в яркости; 2) физическая фотометрия, в которой для сравнения двух источников света используются различные приемники света иного рода – вакуумные фотоэлементы, полупроводниковые фотодиоды и т.д.
32.Закон Бугера-Ламберта-Бера, его использование в количественном анализе.
Физический закон, определяющий ослабление параллельного монохроматического пучка света при распространении его в поглощающей среде.
Закон выражается следующей формулой:
![]() ,
,

где -интенсивность входящего пучка, - толщина слоя вещества, через которое проходит свет,-показатель поглощения (не путать с безразмерным показателем поглощения , который связан сформулой, где- длина волны).
Показатель поглощения характеризует свойства вещества и зависит от длины волны λ поглощаемого света. Эта зависимость называется спектром поглощения вещества.
Для растворов поглощающих веществ в непоглощающих свет растворителях показатель поглощения может быть записан как
где - коэффициент, характеризующий взаимодействиемолекулы поглощающего растворённого вещества со светом с длиной волны λ, -концентрациярастворённого вещества, моль/л.
Утверждение, что не зависит от, называется законом Бера (не путать сзаконом Бэра). Этот закон предполагает, что на способность молекулы поглощать свет не влияют другие окружающие её молекулы этого же вещества в растворе. Однако, наблюдаются многочисленные отклонения от этого закона, особенно при больших .
Если через некоторый слой раствора или газа толщиной (проходит световой поток интенсивностью I, то по закону Ламберта - Бера количество поглощенного света будет пропорционально интенсивности /, концентрации с вещества, поглощающего свет, и толщине СЛОЯ) закон БМБ, который связывает интенсивности света, падающего на вещество и прошедшего его, с концентрацией вещества и толщиной поглощающего слоя Ну это так же, как преломление, только затухание в веществе. Которое свет поглощает под определенным процентом. То есть остаток от выхода света есть
33.ИК-спектроскопия.
Этот метод анализа основан на записи инфракрасных спектров поглощения вещества. Поглощение веществом в области инфракрасного излучения происходят за счёт так колебаний атомов в молекулах. Колебания подразделяются на валентные (когда в ходе колебания изменяются расстояния между атомами) и колебательные (когда в ходе колебания изменяются углы между связями). Переходы между различными колебательными состояниями в молекулах квантованы, благодаря чему поглощение в ИК-области имеет форму спектра, где каждому колебанию соответствует своя длина волны. Понятно что длина волны для каждого колебания зависит от того какие атомы в нём участвуют, и кроме того она мало зависит от их окружения.
метод ИК-спектроскопии не являете разделяющим методом, то есть при исследовании какого-либо вещества может оказаться что исследовалась на самом деле смесь нескольких веществ, что конечно сильно исказит результаты расшифровки спектра. Ну и всё ж говорить об однозначной идентификации вещества с помощью метода ИК-спектроскопии не вполне правильно, так как метод скорее позволяет выявить определённые функциональные группы, а не их количество в соединении и их способ связи друг с другом.
метод ИК-спектроскопии используется при проведении исследований полимерных материалов, волокон, лакокрасочных покрытий, наркотических средств (при идентификации наполнителя в качестве которого часто выступают углеводы в том числе полисахариды). Особенно метод незаменим при исследовании смазочных материалов, тем что даёт возможность одновременного определения природы как основы смазочного материала, так и возможных добавок (присадок) к этой основе.
34. Рентгенофлуоресцентный анализ.
(РФА) - один из современных спектроскопических методов исследования вещества с целью получения его элементного состава, то есть его элементного анализа. С помощью него могут анализироваться различные элементы от бериллия (Be) до урана (U). Метод РФА основан на сборе и последующем анализе спектра, полученного путём воздействия на исследуемый материал рентгеновским излучением. При облучении атом переходит в возбуждённое состояние, заключающееся в переходе электронов на более высокие энергетические уровни. В возбуждённом состоянии атом пребывает крайне малое время, порядка одной микросекунды, после чего возвращается в спокойное положение (основное состояние). При этом электроны с внешних оболочек либо заполняют образовавшиеся вакантные места, а излишек энергии испускается в виде фотона, либо энергия передается другому электрону из внешних оболочек (оже-электрон)
Экология и охрана окружающей среды: определение тяжёлых металлов в почвах, осадках, воде, аэрозолях и др.
Геология и минералогия: качественный и количественный анализ почв, минералов, горных пород и др.
Металлургия и химическая индустрия: контроль качества сырья, производственного процесса и готовой продукции
Лакокрасочная промышленность: анализ свинцовых красок
35. Атомно-эмиссионная спектроскопия.
Атомно-эмиссионный спектральный анализ - это совокупность методов элементного анализа, основанных на изучении спектров испускания свободных атомов и ионов в газовой фазе. Обычно эмиссионные спектры регистрируют в наиболее удобной оптической области длин волн от 200 до 1000 нм.
АЭС (атомно-эмиссионная спектрометрия) – способ определения элементного состава вещества по оптическим спектрам излучения атомов и ионов анализируемой пробы, возбуждаемым в источниках света. В качестве источников света для атомно-эмиссионного анализа используют пламя горелки или различные виды плазмы, включая плазму электрической искры или дуги, плазму лазерной искры, индуктивно-связанную плазму, тлеющий разряд и др. АЭС – самый распространённый экспрессный высокочувствительный метод идентификации и количественного определения элементов примесей в газообразных, жидких и твердых веществах, в том числе и в высокочистых.
Области применения:
Металлургия: анализ состава металлов и сплавов,
Горнодобывающая промышленность: исследование геологических образцов и минерального сырья,
Экология: анализ воды и почвы,
Техника: анализ моторных масел и др. технических жидкостей на примеси металлов,
Биологические и медицинские исследования.
Принцип действия.
Принцип действия атомно-эмиссионного спектрометра достаточно прост. Он основан на том, что атомы каждого элемента могут испускать свет определенных длин волн - спектральные линии, причем эти длины волн разные для разных элементов. Для того чтобы атомы начали испускать свет, их необходимо возбудить – нагреванием, электрическим разрядом, лазером или каким-либо иным способом. Чем больше атомов данного элемента присутствует в анализируемом образце, тем ярче будет излучение соответствующей длины волны.
Интенсивность спектральной линии анализируемого элемента, помимо концентрации анализируемого элемента, зависит от большого числа различных факторов. По этой причине рассчитать теоретически связь между интенсивностью линии и концентрацией соответствующего элемента невозможно. Вот почему для проведения анализа необходимы стандартные образцы, близкие по составу к анализируемой пробе. Предварительно эти стандартные образцы экспонируются (прожигаются) на приборе. По результатам этих прожигов для каждого анализируемого элемента строится градуировочный график, т.е. зависимость интенсивности спектральной линии элемента от его концентрации. Впоследствии, при проведении анализа проб, по этим градуировочным графикам и производится пересчет измеренных интенсивностей в концентрации.
Подготовка проб для анализа.
Следует иметь виду, что реально анализу подвергается несколько миллиграммов пробы с ее поверхности. Поэтому для получения правильных результатов проба должна быть однородна по составу и структуре, при этом состав пробы должен быть идентичным составу анализируемого металла. При анализе металла в литейном или плавильном производстве для отливки проб рекомендуется использовать специальные кокили. При этом форма пробы может быть произвольной. Необходимо лишь, чтобы анализируемый образец имел достаточную поверхность и мог быть зажат в штативе. Для анализа мелких образцов, например прутков или проволоки, могут быть использованы специальные адаптеры.
Преимущества метода:
Бесконтактность,
Возможность одновременного количественного определения большого числа элементов,
Высокая точность,
Низкие пределы обнаружения,
Простота пробоподготовки,
Низкая себестоимость.
36. Атомно-абсорбционная спектроскопия.
метод количеств.определения элементного состава исследуемого вещества по атомным спектрам поглощения, основанныйна способности атомов избирательно поглощать электромагнитное излучение в разл. участках спектра. A.-a.a. проводят на спец. приборах - абсорбц. спектрофотометрах. Пробу анализируемого материала растворяют(обычно c образованием солей); раствор в виде аэрозоля подают в пламя горелки. Под действием пламени(3000°C) молекулы солей диссоциируют на атомы, к-рые могут поглощать свет. Затем через пламя горелкипропускают пучок света, в спектре к-рого есть соответствующие тому или иному элементу спектральныелинии. Из общего излучения исследуемые спектральные линии выделяют монохроматором, a ихинтенсивность фиксируют блоком регистрации. Mатем. обработка проводится по формуле: J = J0 * e-kvI,
где J и J0, - интенсивности прошедшего и падающего света; kv – коэфф. поглощения, зависящий от егочастоты; I - толщина поглощающего слоя
более чувствительный чем АЭС
37. Нефелометрия и турбидиметрия.
S = lg (I°/I) интенсивность падающ. В р-р(I°) делим на интенсивность выходщ из р-ра(I) =
k-const мутности
b – длина пути пучка света
N-число частиц в ед. р-ра
В нефелометрическом и турбидиметрическом анализе используется явление рассеяния света твердыми частицами, находящимися в растворе во взвешенном состоянии.
Нефелометрия - метод определения дисперсности и концентрации коллоидных систем по интенсивности рассеянного ими света. Нефелометрия, измерения производятся в специальном приборе нефелометре, действие которого основано на сравнении интенсивности рассеянного исследуемой средой света с интенсивностью света, рассеянного другой средой, служащей стандартом. Теория рассеяния света коллоидными системами, в которых размеры частиц не превышают длины полуволны падающего света, была разработана английским физиком Дж. Рэлеем в 1871. По закону Рэлея, интенсивность света I, рассеянного в направлении, перпендикулярном к падающему лучу, выражается формулой I=QNvlk -где q- интенсивность падающего света, N - общее число частиц в единице объёма, или частичная концентрация, v - объём одной частицы, \ - длина волны падающего света, k - константа, зависящая от показателей преломления коллоидных частиц и окружающей их дисперсионной среды, расстояния от источника света, а также от принятых единиц измерения
Турбидиметрия - метод анализа мутных сред, основанный на измерении интенсивности поглощенного ими света. Турбидиметрические измерения производят в проходящем свете с помощью турбидиметров визуальных или фотоэлектрических колориметров. Методика измерений аналогична колориметрической и основывается на применимости к мутным средам Бугера -Ламберта - закона Бэра, который в случае суспензий справедлив лишь для очень тонких слоев или при значительных разбавлениях. При турбидиметрии требуется тщательное соблюдение условий образования дисперсной фазы, аналогичных условиям, соблюдаемым при нефелометрии. Значительное усовершенствование турбидиметрии заключается в применении турбидиметрического титрования по максимуму помутнения с помощью фотоэлектрических колориметров. Турбидиметрия с успехом используются для аналитического определения сульфатов, фосфатов, хлоридов, цианидов, свинца, цинка и др.
Основным достоинством нефелометрических и турбидиметрических методов является их высокая чувствительность, что особенно ценно по отношению к элементам или ионам, для которых отсутствуют цветные реакции. В практике широко применяется, например, нефелометрическое определение хлорида и сульфата в природных водах и аналогичных объектах. По точности турбидиметрия и нефелометрия уступают фотометрическим методам, что связано, главным образом, с трудностями получения суспензий, обладающих одинаковыми размерами частиц, стабильностью во времени и т. д. К обычным сравнительно небольшим погрешностям фотометрического определения добавляются ошибки, связанные с недостаточной воспроизводимостью химико-аналитических свойств суспензий.
Нефелометрию и турбидиметрию применяют, напр., для определения SO4 в виде взвеси BaSO4, Сl- в виде взвеси AgCl, S2- в виде взвеси CuS с ниж. границами определяемых содержаний ~ 0,1 мкг/мл. Для стандартизации условий анализа в экспериментах необходимо строго контролировать т-ру, объем взвеси, концентрации реагентов, скорость перемешивания, время проведения измерений. Осаждение должно протекать быстро, а осаждающиеся частицы должны иметь малые размеры и низкую р-римость. Для предотвращения коагуляции крупных частиц в р-р часто добавляют стабилизатор, напр. желатин, глицерин.
38. Хроматография: история возникновения, принцип метода, применение в суд. Исследованиях.
Хроматогра́фия- динамический сорбционный метод разделения и анализа смесей веществ, а также изучения физико-химических свойств веществ. Основан на распределении веществ между двумя фазами - неподвижной (твердая фаза или жидкость, связанная на инертном носителе) и подвижной (газовая или жидкая фаза, элюент). Название метода связано с первыми экспериментами по хроматографии, в ходе которых разработчик метода Михаил Цвет разделял ярко окрашенные растительные пигменты.
Метод хроматографии был впервые применён русским учёным-ботаником Михаилом Семеновичем Цветом в 1900 году. Он использовал колонку, заполненнуюкарбонатом кальция, для разделения пигментов растительного происхождения. Первое сообщение о разработке метода хроматографии было сделано Цветом 30 декабря 1901 года на XI Съезде естествоиспытателей и врачей в С.-Петербурге. Первая печатная работа по хроматографии была опубликована в 1903 году, в журнале Труды Варшавского общества естествоиспытателей . Впервые термин хроматография появился в двух печатных работах Цвета в 1906 году, опубликованных в немецком журнале Berichte der Deutschen Botanischen Gesellschaft . В 1907 году Цвет демонстрирует свой метод Немецкому Ботаническому обществу .
В 1910-1930 годы метод был незаслуженно забыт и практически не развивался.
В 1931 году Р. Кун, А. Винтерштейн и Е. Ледерер при помощи хроматографии выделили из сырого каротина α и β фракции в кристаллическом виде, чем продемонстрировали препаративную ценность метода.
В 1941 году А. Дж. П. Мартин и Р. Л. М. Синг разработали новую разновидность хроматографии, в основу которой легло различие в коэффициентах распределения разделяемых веществ между двумя несмешивающимися жидкостями. Метод получил название «распределительная хроматография ».
В 1947 году Т. Б. Гапон, Е. Н. Гапон и Ф. М. Шемякин разработали метод «ионообменной хроматографии».
В 1952 году Дж. Мартину и Р. Сингу была присуждена Нобелевская премия в области химии за создание метода распределительной хроматографии.
С середины XX века и до наших дней хроматография интенсивно развивалась и стала одним из наиболее широко применяемых аналитических методов.
Классификация: Газовая, Жидкостная
Основы хроматографич. процесса. Для проведения хроматографич. разделения в-в или определения их физ.-хим. характеристик обычно используют спец. приборы - хроматографы. Осн. узлы хроматографа - хроматографич. колонка, детектор, а также устройство для ввода пробы. Колонка, содержащая сорбент, выполняет ф-цию разделения анализируемой смеси на составные компоненты, а детектор -ф-цию их количеств. определения. Детектор, расположенный на выходе из колонки, автоматически непрерывно определяет концентрацию разделяемых соед. в потоке подвижной После ввода анализируемой смеси с потоком подвижной фазы в колонку зоны всех в-в расположены в начале хроматографич. колонки (рис. 1). Под действием потока подвижной фазы компоненты смеси начинают перемещаться вдоль колонки с разл. скоростями, величины к-рых обратно пропорциональны коэффициентам распределения К хроматографируемых компонентов. Хорошо сорбируемые в-ва, значения константраспределения для к-рых велики, передвигаются вдоль слоя сорбента по колонке медленнее, чем плохо сорбируемые. Поэтому быстрее всех из колонки выходит компонент А, затем компонент Б и последним покидает колонку компонент В (К А <К Б <К В). Сигнал детектора, величина к-рого пропорциональна концентрации определяемого в-ва в потоке элюента, автоматически непрерывно записывается и регистрируется (напр., на диаграммной ленте). Полученная хроматограмма отражает расположение хроматографич. зон на слое сорбента или в потоке подвижной фазы во времени.

Рис. 1. Разделение смеси из трех компонентов (А, Б и В) на хроматографической колонке К с детектором Д: а - положение хроматографических зон разделяемых компонентов в колонке через определенные интервалы времени; б - хроматограмма (С - сигнал, t - время).
При плоскослойном хроматографич. разделении лист бумаги или пластину со слоем сорбента с нанесеннымипробами исследуемого в-ва помещают в хроматографич. камеру. После разделения компоненты определяют любым подходящим методом.
39. Классификация хроматографических методов.
Храмотография – метод разделения и анализа веществ, основанный на распределении анализир. В-ва между 2 фазами: подвижной и неподвижной
Раствор смеси веществ подлежащих разделению, пропускают через стеклянную трубку(Адсорбционную колонку) заполненную адсорбентом. В результате компоненты смеси удерживаются на различной высоте столба адсорбента в виде отдельных зон (слоев). Вещ-ва лучше адсорбир. Нах в верх части столба, а хуже адсорбируемые в ниж части столба. В-ва не способные адсорбироваться - проходят через колонку не задерживаясь и собираются в фильтре.
Классификации:
1. По агрегатному состоянию фаз.
1) Подвижная
А)газовая (инертные газы:гелий,аргон,азон)
Б)жидкостная
2. по способу проведения
1) на плоскости(планарная); бумажная тонкослойная
2) колоночная
А) насадочная(насадочная колонка наполненная сорбентом)
Б) капиллярная (тонкий стеклянный/кварцевый капиляр на внутр.поверхности которого нанесена неподвижная фаза)
Можно опр. Вещ-ва в небольш.кол-вах.
Летучие в-ва разделяются.
40. Хроматограмма. Основные параметры хроматограф.пика.
Хроматограмма - результат регистрирования зависимости концентрации компонентов на выходе из колонки от времени.
H S
Каждый пик на хроматограмме характеризуется двумя основными параметрами
1. Время удерживания (t R ) – это время от момента ввода анализируемой пробы до момента регистрации максимума хроматографического пика. Оно зависит от природы вещества и является качественной характеристикой.
2. Высота (h ) или площадь (S ) пика
S = ½ ω × h . (4)
Высота и площадь пика зависят от количества вещества и являются количественными характеристиками.
Время удерживания складывается из двух составляющих – времени пребывания веществ в подвижной фазе (t m ) и времени пребывания в неподвижной фазе (t s ):
Идентификацию пиков неизвестных компонентов анализируемой смеси проводят путем сопоставления (сравнения) относит. величин, определяемых непосредственно из хроматограммы, с соответствующими табличными данными для известных соединений. При идентификации в хроматографии достоверен только отрицат. ответ; напр., пик i не является в-вом А, если времена удерживания пика i и в-ва А не совпадают. Совпадение времен удерживания пика i и в-ва А - необходимое, но недостаточное условие для заключения, что пик i - это в-во А.
В практической работе выбор того или иного параметра для количественной расшифровки хроматограмм определяется совокупным влиянием нескольких факторов быстротой и удобством расчета, формой (широкий, узкий) и степенью асимметрии хроматографического пика, эффективностью используемой колонки, полнотой разделения компонентов смеси, наличием необходимых автоматизированных устройств (интеграторов, компьютерных систем обработки данных хроматографического аиализа).
Определяемый параметр хроматографического пика измеряется оператором на хроматограмме вручную по окончании цикла разделения компонентов анализируемой смеси
Определяемый параметр хроматографического пика измеряется автоматически с помощью цифровых вольтметров, интеграторов или специализированных ЭВМ одновременно с разделением компонентов анализируемой смеси в колонке и записью хроматограммы
Поскольку техника расшифровки хроматограмм сводится к измерению параметров хроматографических пиков интересующего и стандартного соединений, условия хроматографирования должны обеспечивать по возможности полное их разделение все остальные составляющие исходной пробы в принятых условиях анализа могут не отделяться друг от друга или даже вообще не проявляться на хроматограмме (в этом заключается преимущество метода внутреннего стандарта перед методом внутренней нормализации)
41.Качественный хроматографич.анализ.
При достаточной длине колонки можно произвести полное разделение компонентов любой смеси. А после элюирования разделенных компонентов в отдельные фракции (элюаты) определить количество компонентов смеси (оно соответствует количеству элюатов), установить их качественный состав, определить количество каждого из них, использовав соответствующие методы количественного анализа.
Качественный хроматографический анализ, т.е. индетификация вещества по его хроматограмме, может быть выполнен сравнением хроматограических характеристик, чаще всего удерживаемого объема (т.е. объема подвижной фазы, пропущенной через колонку от начала ввода смеси до появления данного компонента на выходе из колонки), найденных при определенных условиях для компонентов анализируемой смеси и для эталона.
42.Количественный хроматограф.анализ.
Количественный хроматографический анализ проводят обычно на хроматографе. Метод основан на измерении различных параметров хроматографического пика, зависящих от концентрации хроматографируемых веществ – высоты, ширины, площади и удерживаемого объема или произведения удерживаемого объема на высоту пика.
В количественной газовой хроматографии применяют методы абсолютной градуировки и внутренней нормализации, или нормировки. Используется также метод внутреннего стандарта. При абсолютной градуировке экспериментально определяют зависимость высоты или площади пика от концентрации вещества и строят градуировочные графики или рассчитывают соответствующие коэффициенты. Далее определяют те же характеристики пиков в анализируемой смеси, и по градуировочному графику находят концентрацию анализируемого вещества. Этот простой и точный метод является основным при определении микропримесей.
При использовании метода внутренней нормализации принимают сумму каких-либо параметров пиков, например, сумму высот всех пиков или сумму их площадей, за 100%. Тогда отношение высоты отдельного пика к сумме высот или отношение площади одного пика к сумме площадей при умножении на 100 будет характеризовать массовую долю (%) компонента в смеси. При таком подходе необходимо, чтобы зависимость величины измеряемого параметра от концентрации была одинаковой для всех компонентов смеси.
43.Планарная хроматография. Использование тонкослойной хроматографии для анализа чернил.
Первой формой использования целлюлозы в тонкослойной хроматографии была бумажная хро-матография. Доступные пластинки для ТСХ и высокопроизводительной ТСХ позволяют разделять смеси полярных веществ, при этом в качестве элюента используются, по крайней мере, тройные смеси из воды, несмешивающегося с ней органического растворителя и водорастворимого рас-творителя, способствующего образованию одной фазы}